Compare Sequences Using Sequence Alignment Algorithms
Determining the similarity between two sequences is a common task in computational biology. Starting with a nucleotide sequence for a human gene, this example uses alignment algorithms to locate and verify a corresponding gene in a model organism.
In this example, you are interested in studying Tay-Sachs disease. Tay-Sachs is an autosomal recessive disease caused by the absence of the enzyme beta-hexosaminidase A (Hex A). This enzyme is responsible for the breakdown of gangliosides (GM2) in brain and nerve cells.
First, research information about Tay-Sachs and the enzyme that is associated with this disease, then find the nucleotide sequence for the human gene that codes for the enzyme, and finally find a corresponding gene in another organism to use as a model for study.
In the MATLAB Command window, enter:
web('https://www.ncbi.nlm.nih.gov/books/NBK22250/')
The system web browser opens with the Tay-Sachs disease page in the Genes and Diseases section of the NCBI web site. This section provides a comprehensive introduction to medical genetics. In particular, this page contains an introduction and pictorial representation of the enzyme Hex A and its role in the metabolism of the lipid GM2 ganglioside.
After completing your research, you have concluded the following:
The gene HEXA codes for the alpha subunit of the dimer enzyme hexosaminidase A (Hex A), while the gene HEXB codes for the beta subunit of the enzyme. A third gene, GM2A, codes for the activator protein GM2. However, it is a mutation in the gene HEXA that causes Tay-Sachs.
Retrieve Sequence Information from a Public Database
The following procedure illustrates how to find the nucleotide sequence for a human gene in a public database and read the sequence information into the MATLAB environment. Many public databases for nucleotide sequences (for example, GenBank®, EMBL-EBI) are accessible from the Web.
After you locate a sequence, you need to move the sequence data into the MATLAB Workspace.
In the MATLAB Command Window, enter:
web('https://www.ncbi.nlm.nih.gov/')
Search for the gene you are interested in studying. For example, from the Search list, select Nucleotide, and in the for box enter Tay-Sachs. Look for the genes that code the alpha and beta subunits of the enzyme hexosaminidase A (Hex A), and the gene that codes the activator enzyme. The NCBI reference for the human gene HEXA has accession number NM_000520.
Get sequence data into the MATLAB environment. For example, to get sequence information for the human gene HEXA, enter:
humanHEXA = getgenbank('NM_000520')humanHEXA = struct with fields:
LocusName: 'NM_000520'
LocusSequenceLength: '4785'
LocusNumberofStrands: ''
LocusTopology: 'linear'
LocusMoleculeType: 'mRNA'
LocusGenBankDivision: 'PRI'
LocusModificationDate: '05-OCT-2024'
Definition: 'Homo sapiens hexosaminidase subunit alpha (HEXA), transcript variant 2, mRNA.'
Accession: 'NM_000520'
Version: 'NM_000520.6'
GI: ''
Project: []
DBLink: []
Keywords: 'RefSeq; MANE Select.'
Segment: []
Source: 'Homo sapiens (human)'
SourceOrganism: [4×65 char]
Reference: {[1×1 struct] [1×1 struct] [1×1 struct] [1×1 struct] [1×1 struct] [1×1 struct] [1×1 struct] [1×1 struct] [1×1 struct] [1×1 struct]}
Comment: [46×66 char]
Features: [160×74 char]
CDS: [1×1 struct]
Sequence: 'ctcacgtggccagccccctccgagaggggagaccagcgggccatgacaagctccaggctttggttttcgctgctgctggcggcagcgttcgcaggacgggcgacggccctctggccctggcctcagaacttccaaacctccgaccagcgctacgtcctttacccgaacaactttcaattccagtacgatgtcagctcggccgcgcagcccggctgctcagtcctcgacgaggccttccagcgctatcgtgacctgcttttcggttccgggtcttggccccgtccttacctcacagggaaacggcatacactggagaagaatgtgttggttgtctctgtagtcacacctggatgtaaccagcttcctactttggagtcagtggagaattataccctgaccataaatgatgaccagtgtttactcctctctgagactgtctggggagctctccgaggtctggagacttttagccagcttgtttggaaatctgctgagggcacattctttatcaacaagactgagattgaggactttccccgctttcctcaccggggcttgctgttggatacatctcgccattacctgccactctctagcatcctggacactctggatgtcatggcgtacaataaattgaacgtgttccactggcatctggtagatgatccttccttcccatatgagagcttcacttttccagagctcatgagaaaggggtcctacaaccctgtcacccacatctacacagcacaggatgtgaaggaggtcattgaatacgcacggctccggggtatccgtgtgcttgcagagtttgacactcctggccacactttgtcctggggaccaggtatccctggattactgactccttgctactctgggtctgagccctctggcacctttggaccagtgaatcccagtctcaataatacctatgagttcatgagcacattcttcttagaagtcagctctgtcttcccagatttttatcttcatcttggaggagatgaggttgatttcacctgctggaagtccaacccagagatccaggactttatgaggaagaaaggcttcggtgaggacttcaagcagctggagtccttctacatccagacgctgctggacatcgtctcttcttatggcaagggctatgtggtgtggcaggaggtgtttgataataaagtaaagattcagccagacacaatcatacaggtgtggcgagaggatattccagtgaactatatgaaggagctggaactggtcaccaaggccggcttccgggcccttctctctgccccctggtacctgaaccgtatatcctatggccctgactggaaggatttctacatagtggaacccctggcatttgaaggtacccctgagcagaaggctctggtgattggtggagaggcttgtatgtggggagaatatgtggacaacacaaacctggtccccaggctctggcccagagcaggggctgttgccgaaaggctgtggagcaacaagttgacatctgacctgacatttgcctatgaacgtttgtcacacttccgctgtgaattgctgaggcgaggtgtccaggcccaacccctcaatgtaggcttctgtgagcaggagtttgaacagacctgagccccaggcaccgaggagggtgctggctgtaggtgaatggtagtggagccaggcttccactgcatcctggccaggggacggagccccttgccttcgtgccccttgcctgcgtgcccctgtgcttggagagaaaggggccggtgctggcgctcgcattcaataaagagtaatgtggcatttttctataataaacatggattacctgtgtttaaaaaaaaaagtgtgaatggcgttagggtaagggcacagccaggctggagtcagtgtctgcccctgaggtcttttaagttgagggctgggaatgaaacctatagcctttgtgctgttctgccttgcctgtgagctatgtcactcccctcccactcctgaccatattccagacacctgccctaatcctcagcctgctcacttcacttctgcattatatctccaaggcgttggtatatggaaaaagatgtaggggcttggaggtgttctggacagtggggagggctccagacccaacctggtcacagaagagcctctcccccatgcatactcatccacctccctcccctagagctattctcctttgggtttcttgctgcttcaattttatacaaccattatttaaatattattaaacacatattgttctctaggcactgtggtagtgggtttttttgttgtttttgtttttgagactgtctcaaaaactctgtcgcccaggctgacagtgcagtggcacaatcttggctcactgcagcctctgcctcctgggttcaagcgattctcgtgcatcagcctcctgagtaactggaattaataggcacgtgccaccatgtccatctaattcatatatatatattttttttttctgagacggagtctcactgtcacacaggctggagtgcagtggcacgatctcgactcactgcaagctccacctcctgggttcacgccattctcctgcctcagcctccccagtagctgggactacaggcgcccgccaccacgcccggctaattttttgtatttttagtagagatggggtttctccgtgttagccaggatggtctcgatctcctgacctcgtgatccgcccgccttggcctcccaaagtgctgggattacaggcgtgagccaccgcgcccggccgaattcatctatttttagtagagatggggtttcactatattggccaggctggtcttgaactcatgacctcagatgttcacttgtcttggcctcccaaagtgctgggattagaggcgtgagccaccgcacgcgggcctgtggtaaattgttgaatttgaaggactcagaggccctggtcaattccaaaataacgtaggcgacttccatccccctcctcccaaccattttcagcccaaagcatcttcgcagggaatggatggctgcgcggaggtgggcggtggctctggagagggtctttgcaggtgtgattttctctagaaggaaatgtctcgtcgtggacccagactgccccctcctggtttcagatgcagaagtgatactgtaagccagaggcgggggcagtaatgcatcgcagccattttaggtgaggatttccttggcggttatttgttaagttctttggctgggccctgggctggggtaacaatggacaggttccaggcatttttttcagaaagcttccagtgtagtggatacagaaacttcaggaaggcagggctgagaaggatctgagtaaaactcggtccttcaacaccatccttcagcccctgggtcatgttccttcgaggtcctggtgggaggtagacaagcctagccttgtgctgttcctgtaaggacagggtgggcattttctaccaacagaattcttggaattttcacacagcccagcctagccaagtccagggctatagcccagatacacaagttaaggtcccagcactggcacccaccacaggagcccccttacctctattacccagaagcttgtaggaggggtggtccgcagacaaggaccctgcacaggtgcgaccctgcttccctcctggtcataactttcatgttactattgcttgggataatgttaagtaaaaatagcagacactgagttttaagtctcaagtggatgaaggcagagatcgtgatacacttgagttaaagcagtagggttctgtcattttctattcctgttgtaaacattttctttaatgttattatttttaccactaaactaacgtggcctggtcacgactttcattggtaaagtgtgctgttcctcaccctccaccgttgctcctttggtccactgatcataagagcatttacctgaaggtcgtcagacctcgaatgccaacaggtcaactgcagtggcctgcagttaccacccagtctgttccaatgaacagaatcgctgttgccccaactcatctcccttcacctaggctgtaaattgaaagtcccacccctgagcggaacacaggccatcttgtgtgctgtgcaccaccagggggtggggaagtttccagactgacttcctggctccagtcatcctaggaaaagagttctccagtcgctccccacccccaccccttcccattccaggagtctattaaggaggcaaagcaggcctaacgggtatcaaagcaaaggagtgaatggagactgggagagtcttcaacctctcctctccttggtaggagctgaggctgcatgccaggtaccttcccttcgaggaatctaataaagctaggtcactggtgttttcaggtgcttctcaaaggattgccgtaggggtaggatatcaggatgtgggagcacaggtgccaccacagcactagtgatggagagtcattgcccctagacttctgggacagtgagactgtgaggaaagctgaaatgatactgggaaagggtgaaagaaaggatgtaggtggaatttatttagtattaatgtaggtacacataccttatggcaacattcctagcactctaaattctagatttgtatagtttctgtcaatatcttttgtaagcttaatcaatacagggcatgacaagtatgtgtcacatacttttttttccacgaagaaaaaaaataagtaggaattgggtgctttgtttatcaaaatttgtatttcctttataaataaactttgaaataaaggttgaaaattagta'
SearchURL: 'https://www.ncbi.nlm.nih.gov/entrez/viewer.fcgi?db=nuccore&id=NM_000520'
RetrieveURL: 'https://eutils.ncbi.nlm.nih.gov/entrez/eutils/efetch.fcgi?db=nuccore&id=1677538638&rettype=gb&retmode=text&api_key=55022f38eb25e2f6b00a772015c7b77d6208'
Search a Public Database for Related Genes
The following procedure illustrates how to find the nucleotide sequence for a mouse gene related to a human gene, and read the sequence information into the MATLAB environment. The sequence and function of many genes is conserved during the evolution of species through homologous genes. Homologous genes are genes that have a common ancestor and similar sequences. One goal of searching a public database is to find similar genes. If you are able to locate a sequence in a database that is similar to your unknown gene or protein, it is likely that the function and characteristics of the known and unknown genes are the same.
After finding the nucleotide sequence for a human gene, you can do a BLAST search or search in the genome of another organism for the corresponding gene. This procedure uses the mouse genome as an example.
In the MATLAB Command window, enter:
web('http://www.ncbi.nlm.nih.gov')
Search the nucleotide database for the gene or protein you are interested in studying. For example, from the Search list, select Nucleotide, and in the for box enter hexosaminidase A.
The search returns entries for the mouse and human genomes. For the purposes of this example, use the accession number AK080777 for the mouse gene HEXA.
Get sequence information for the mouse gene into the MATLAB environment.
mouseHEXA = getgenbank('AK080777')
Compare Amino Acid Sequences
The following procedure illustrates how to use global and local alignment functions to compare two amino acid sequences. You could use alignment functions to look for similarities between two nucleotide sequences, but alignment functions return more biologically meaningful results when you are using amino acid sequences.
If you did not retrieve gene data from the Web, you can load example data from a MAT-file included with the Bioinformatics Toolbox™ software. In the MATLAB Command window, enter:
load hexosaminidaseConvert the human and mouse DNA sequences to the amino acid sequences.
humanProtein = nt2aa(humanHEXA.Sequence); mouseProtein = nt2aa(mouseHEXA.Sequence);
Draw a dot plot comparing the human and mouse amino acid sequences. Dot plots are one of the easiest ways to look for similarity between sequences. The diagonal line shown below indicates that there may be a good alignment between the two sequences.
warning('off','bioinfo:seqdotplot:imageTooBigForScreen'); seqdotplot(mouseProtein,humanProtein,4,3); ylabel('Mouse hexosaminidase A (alpha subunit)') xlabel('Human hexosaminidase A (alpha subunit)') uif = gcf; uif.Position(:) = [100 100 1280 800]; % Resize the figure.
warning('on','bioinfo:seqdotplot:imageTooBigForScreen');
Globally align the two amino acid sequences, using the Needleman-Wunsch algorithm.
[GlobalScore, GlobalAlignment] = nwalign(humanProtein,mouseProtein)
GlobalScore = 634.3333
GlobalAlignment = 3×812 char array
'SCRRPAQSAARSRSLRSRPEVKGQGVGPPGVAGAEPPLVT*FADKSRGRRSPDQGLTWPAPSERGDQRAMTSSRLWFSLLLAAAFAGRATALWPWPQNFQTSDQRYVLYPNNFQFQYDVSSAAQPGCSVLDEAFQRYRDLLFGSGSWPRPYLTGKRHTLEKNVLVVSVVTPGCNQLPTLESVENYTLTINDDQCLLLSETVWGALRGLETFSQLVWKSAEGTFFINKTEIEDFPRFPHRGLLLDTSRHYLPLSSILDTLDVMAYNKLNVFHWHLVDDPSFPYESFTFPELMRKGSYNPVTHIYTAQDVKEVIEYARLRGIRVLAEFDTPGHTLSWGPGIPGLLTPCYSGSEPSGTFGPVNPSLNNTYEFMSTFFLEVSSVFPDFYLHLGGDEVDFTCWKSNPEIQDFMRKKGFGEDFKQLESFYIQTLLDIVSSYGKGYVVWQEVFDNKVKIQPDTIIQVWREDIPVNYMKELELVTKAGFRALLSAPWYLNRISYGPDWKDFYIVEPLAFEGTPEQKALVIGGEACMWGEYVDNTNLVPRLWPRAGAVAERLWSNKLTSDLTFAYERLSHFRCELLRRGVQAQPLNVGFCEQEFEQT*APGTEEGAGCR*MVVEPGFHCILARGRSPLPSCPLPACPCAWRERGRCWRSHSIKSNVAFFYNKHGLPVFKKKSVNGVRVRAQPGWSQCLPLRSFKLRAGNETYSLCAVLPCL*AMSLPSHS*PYSRHLP*SSACSLHFCIISPRRWYMEKDVGAWRCSGQWGGLQTQPGHRRASPPCILIHLPPLELFSFGFLAASILYNHYLNIIKHILFS'
' || |: | | | | | ||:: ||| |||||||:| ||||||||| :|| :||:||||||||:| |||||| || ||||||:|||:||||||||||| :::|::|| ||:||||||| ||::|:|||||||||||||||||| |||||||||||||||||||||||||||||||:|:|||||||||:|||||||||||||||||||||||||:|||||||||| |||||||||||| ||||:|||||||||||||||||||||||||||||||||||||||||| |||||||||||: ||||||||||||:||:||||:|||:|||||||||||||||||||||||||:|| ||:|||| ||||||||||||||||||:| |||||||||||||||::||||||||||::||:|| |:: :|:|||||||||||||||::|||||||:| ||||||:||||||||||||||||||||||:||||||||||||||||||||::||::: ||::|||||||||:|||:||||::|| |||||||||| | :| : :|| | | || |: :: | | :: | : :| : | :| : : | ::: | | |::| : | | | :| ||::|| | |: | | | :: |: | '
'--------AA------------GR--------G---------A----G-R-------W----------AMAGCRLWVSLLLAAALACLATALWPWPQYIQTYHRRYTLYPNNFQFRYHVSSAAQAGCVVLDEAFRRYRNLLFGSGSWPRPSFSNKQQTLGKNILVVSVVTAECNEFPNLESVENYTLTINDDQCLLASETVWGALRGLETFSQLVWKSAEGTFFINKTKIKDFPRFPHRGVLLDTSRHYLPLSSILDTLDVMAYNKFNVFHWHLVDDSSFPYESFTFPELTRKGSFNPVTHIYTAQDVKEVIEYARLRGIRVLAEFDTPGHTLSWGPGAPGLLTPCYSGSHLSGTFGPVNPSLNSTYDFMSTLFLEISSVFPDFYLHLGGDEVDFTCWKSNPNIQAFMKKKGF-TDFKQLESFYIQTLLDIVSDYDKGYVVWQEVFDNKVKVRPDTIIQVWREEMPVEYMLEMQDITRAGFRALLSAPWYLNRVKYGPDWKDMYKVEPLAFHGTPEQKALVIGGEACMWGEYVDSTNLVPRLWPRAGAVAERLWSSNLTTNIDFAFKRLSHFRCELVRRGIQAQPISVGCCEQEFEQT*A--T--SA--E----HPG-------G------C----CP---------L-SQ-LR--*A--------P---RR-V--LALR-E----Q-VP--G-Q---G-*SFT---------A-SRPGES---T---P----CP---C--APVT--TEKEAGA----GT--GV--Q---*R-----------------------S-MW-HF-------L--'
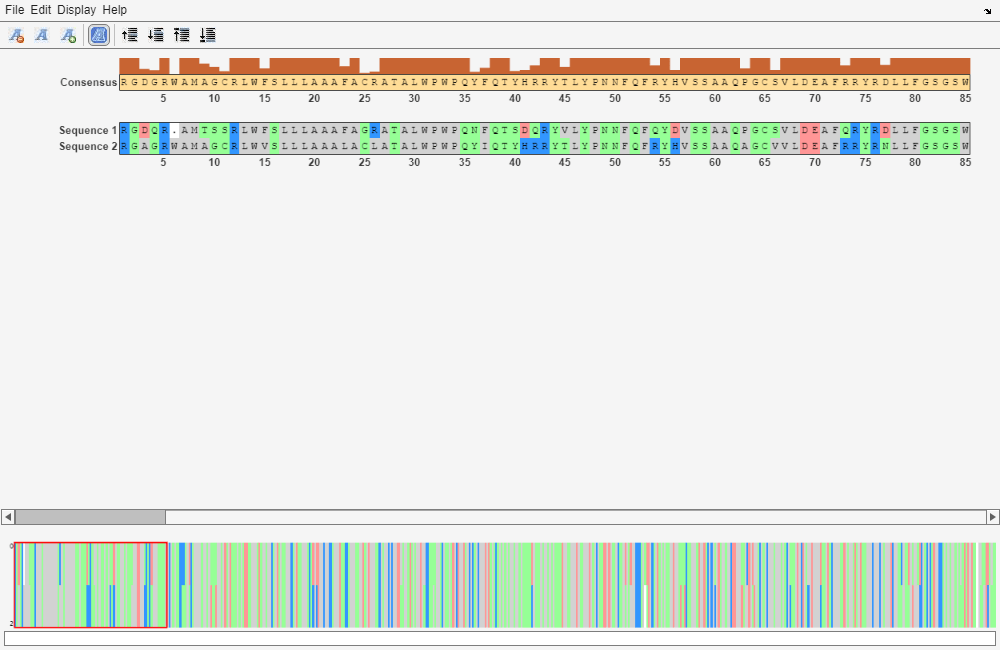
You can also visualize the alignment in the Sequence Alignment app. The alignment is very good between amino acid position 69 and 599, after which the two sequences appear to be unrelated. Notice that there is a stop (*) in the sequence at this point. If you shorten the sequences to include only the amino acids that are in the protein you might get a better alignment. Include the amino acid positions from the first methionine (M) to the first stop (*) that occurs after the first methionine.
seqalignviewer(GlobalAlignment);
Trim the sequence from the first start amino acid (usually M) to the first stop (*) and then try alignment again. Find the indices for the stops in the sequences.
humanStops = find(humanProtein == '*')humanStops = 1×6
41 599 611 713 722 730
mouseStops = find(mouseProtein == '*')mouseStops = 1×4
539 557 574 606
Looking at the amino acid sequence for humanProtein, the first M is at position 70, and the first stop after that position is actually the second stop in the sequence (position 599). Looking at the amino acid sequence for mouseProtein, the first M is at position 11, and the first stop after that position is the first stop in the sequence (position 557).
Truncate the sequences to include only amino acids in the protein and the stop.
humanProteinORF = humanProtein(70:humanStops(2))
humanProteinORF = 'MTSSRLWFSLLLAAAFAGRATALWPWPQNFQTSDQRYVLYPNNFQFQYDVSSAAQPGCSVLDEAFQRYRDLLFGSGSWPRPYLTGKRHTLEKNVLVVSVVTPGCNQLPTLESVENYTLTINDDQCLLLSETVWGALRGLETFSQLVWKSAEGTFFINKTEIEDFPRFPHRGLLLDTSRHYLPLSSILDTLDVMAYNKLNVFHWHLVDDPSFPYESFTFPELMRKGSYNPVTHIYTAQDVKEVIEYARLRGIRVLAEFDTPGHTLSWGPGIPGLLTPCYSGSEPSGTFGPVNPSLNNTYEFMSTFFLEVSSVFPDFYLHLGGDEVDFTCWKSNPEIQDFMRKKGFGEDFKQLESFYIQTLLDIVSSYGKGYVVWQEVFDNKVKIQPDTIIQVWREDIPVNYMKELELVTKAGFRALLSAPWYLNRISYGPDWKDFYIVEPLAFEGTPEQKALVIGGEACMWGEYVDNTNLVPRLWPRAGAVAERLWSNKLTSDLTFAYERLSHFRCELLRRGVQAQPLNVGFCEQEFEQT*'
mouseProteinORF = mouseProtein(11:mouseStops(1))
mouseProteinORF = 'MAGCRLWVSLLLAAALACLATALWPWPQYIQTYHRRYTLYPNNFQFRYHVSSAAQAGCVVLDEAFRRYRNLLFGSGSWPRPSFSNKQQTLGKNILVVSVVTAECNEFPNLESVENYTLTINDDQCLLASETVWGALRGLETFSQLVWKSAEGTFFINKTKIKDFPRFPHRGVLLDTSRHYLPLSSILDTLDVMAYNKFNVFHWHLVDDSSFPYESFTFPELTRKGSFNPVTHIYTAQDVKEVIEYARLRGIRVLAEFDTPGHTLSWGPGAPGLLTPCYSGSHLSGTFGPVNPSLNSTYDFMSTLFLEISSVFPDFYLHLGGDEVDFTCWKSNPNIQAFMKKKGFTDFKQLESFYIQTLLDIVSDYDKGYVVWQEVFDNKVKVRPDTIIQVWREEMPVEYMLEMQDITRAGFRALLSAPWYLNRVKYGPDWKDMYKVEPLAFHGTPEQKALVIGGEACMWGEYVDSTNLVPRLWPRAGAVAERLWSSNLTTNIDFAFKRLSHFRCELVRRGIQAQPISVGCCEQEFEQT*'
Globally align the trimmed amino acid sequences.
[GlobalScore_trim, GlobalAlignment_trim] = nwalign(humanProteinORF,mouseProteinORF); seqalignviewer(GlobalAlignment_trim);

An alternative method to working with subsequences is to use a local alignment function with the nontruncated sequences.
Locally align the two amino acid sequences using a Smith-Waterman algorithm.
[LocalScore, LocalAlignment] = swalign(humanProtein,mouseProtein); seqalignviewer(LocalAlignment);
close all;