FeatureCountOptions
Description
A FeatureCountOptions object contains options to compute the
number of reads mapped to genomic features. This object is used as the value of
Options property of the bioinfo.pipeline.block.FeatureCount block.
Creation
Syntax
Description
optionsObj = bioinfo.pipeline.options.FeatureCountOptionsFeatureCountOptions object with default property values.
optionsObj = bioinfo.pipeline.options.FeatureCountOptions(Name=Value)Name is the property name and
Value is the property value. For example, ssOpt =
bioinfo.pipeline.options.FeatureCountOptions(CountFragments=true) specifies to
count reads as pairs of mates.
Properties
Feature type, specified as a character vector or string. This is used to decide what feature to consider from the GTF file. Default is 'exon'.
Attribute type, specified as a character vector or string. This is used to decide what attribute to consider from the GTF file for grouping features into metafeatures and summarizing the read count.
Boolean variable indicating whether to summarize at the metafeature level, specified as true or false.
Default is true, meaning the function groups features into metafeatures and reports the read counts for metafeatures.
Name of file containing aliases of reference names, specified as a character vector or string. The file must be a tab-delimited file where the first column corresponds to the reference names used in the GTF file, and the second column corresponds to the reference names used in the input file(s). The names are case-sensitive. It is necessary to include only the reference names that are different in the GTF file and the input file. The file must contain only one alias term for any reference listed in the input file. By default, the reference names in the GTF file and those in the input files are assumed to be the same.
Boolean variable indicating whether to count reads as fragments, specified as true or false. Paired-end reads must have the same ID for the field QNAME in the input file, and the mutual order of mates is inferred by the appropriate bit in the FLAG field within the input file. Reads that have no valid mate either because the mate is unmapped or filtered out by input criteria are still counted if they satisfy the overlapping criteria.
Default is false, that is, the reads are counted as single-end reads, and their pairing information is ignored.
Strand specificity of the sequencing protocol, specified as 'unstranded' (default), 'stranded', or 'reverse'.
If
'unstranded', the strand of the reads (or fragments) is ignored.If
'stranded', the strand of the reads (or fragments) is considered, and only those having the same strand as the feature they overlap are counted.If
'reverse', the opposite direction of the strand of the reads (or fragments) is considered, and only those having the opposite strand as the feature they overlap are counted.
When counting fragments (paired-end reads), the strand of the first mate is considered as the strand of the whole fragment. The mutual order of mates (first or second) is inferred from the appropriate bit in the FLAG field of the input file.
Minimum number of overlapped bases required to assign a read to a feature, specified as a positive integer. When counting fragments, the sum of the overlaps from each end is used as the minimum number of overlapped bases.
Minimum mapping quality for a given read to be considered for counting, specified as a non-negative integer. This corresponds to the MAPQ field in the input file. If counting fragments, at least one of the read mates must satisfy this criterion in order to be considered for counting.
Boolean variable indicating whether to count reads overlapping multiple features, specified as true or false (default).
If true, a read (or fragment) overlapping multiple features is counted multiple times. During summarization at the metafeature level, a read (or fragment) is counted only once if it overlaps with multiple features belonging to the same metafeature as long as it does not overlap with other metafeaures.
Counting option for reads having multiple mapping locations in the input file, specified as 'primary' (default), 'none', or 'all'.
If
'primary', only the primary alignment of a multi-mapped read is considered. The appropriate bit in the input file is used to identify primary alignments.If
'none', all alignments of a multi-mapped read are ignored. The NH tag is used to identify multi-mapped reads.If
'all', all alignments of a multi-mapped read are considered and counted multiple times.
Boolean variable indicating whether a fragment must have both mates mapped, specified as true or false. Mate mapping information is retrieved from the FLAG field in the input file. Default is false.
Boolean variable indicating whether a fragment must be properly paired, specified as true or false. Mate pairing information is retrieved from the FLAG field in the input file. Default is false.
Boolean variable indicating whether to report features or metafeatures with zero count for every input file in the output table, specified as true or false.
Default is false, that is, only rows with non-zero counts and columns with non-zero counts are included in the output table.
Method to use when assigning a given read to metafeature, specified as 'partial', 'full', 'max', or 'hits'. If 'Summarization' is set to false, then the reads are assigned to features, instead of metafeatures, based on the specified method.
In the following table, R refers to a read or fragment, and M refers to a metafeature.
| Method | Description |
|---|---|
'partial' | R is assigned to M if R overlaps (even partially) only with M. Otherwise R is considered ambiguous. |
'full' | R is assigned to M if R is completely mapped only within M, that is, fully overlapping only M. Otherwise R is considered ambiguous. |
'max' | R is assigned to M if R satisfies the overlapping criteria only with M, or if R satisfies the overlapping criteria with several metafeatures but overlaps fully only with M. |
'hits' | R is assigned to M if R overlaps even partially only M, or if M is the only metafeature with the highest number of features hit by R; otherwise R is considered ambiguous. |
The following schematic diagram and table illustrate the outcome of these methods in conjunction with the 'CountMultiOverlap' name-value pair argument. In the figure, the read refers to a short-read sequence from an input file, and feature A and feature B refers to features listed in a GTF file.
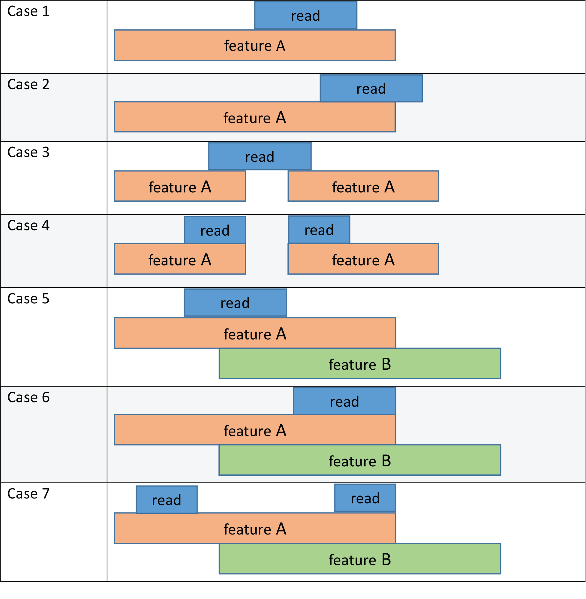
Each method column lists the feature that the read is assigned to based on the corresponding method. The 'CountMultiOverlap' column indicates whether this name-value pair is set to true or false and if it has any effect in the outcome of each method.
'CountMultiOverlap' | 'partial' | 'full' | 'max' | 'hits' | |
|---|---|---|---|---|---|
| Case 1 | No effect since the read maps only to one feature (feature A). | feature A | feature A | feature A | feature A |
| Case 2 | No effect since the read maps only to one feature (feature A). | feature A | no feature | feature A | feature A |
| Case 3 | No effect since the read maps only to one feature (feature A). | feature A | no feature | feature A | feature A |
| Case 4 | No effect since the read maps only to one feature (feature A). | feature A | feature A | feature A | feature A |
| Case 5 | false | ambiguous | feature A | feature A | ambiguous |
true | feature A, feature B | feature A | feature A | feature A, feature B | |
| Case 6 | false | ambiguous | ambiguous | ambiguous | ambiguous |
true | feature A, feature B | feature A, feature B | feature A, feature B | feature A, feature B | |
| Case 7 | false | Ambiguous | feature A | feature A | feature A |
true | feature A, feature B | feature A | feature A | feature A |
no feature means that the read is not assigned to any feature. If you have specified the second output table S, its Unassigned_noFeature row is incremented by one for such occurrence. ambiguous means that the read is not assigned to any feature since it satisfies the overlapping criteria for multiple features, and the Unassigned_ambiguous row is incremented by one for such occurrence.
Boolean variable indicating whether to compute in parallel, specified as
true or false.
In order to execute the computation in parallel, you must have Parallel Computing Toolbox™. If a MATLAB® parallel pool does not exist, one is automatically created when the auto-creation option is enabled in your parallel preferences. Otherwise, computation runs in the serial mode.
Default is false, that is, serial mode.
Boolean variable indicating whether to display the progress of computation, specified as true or false.
Version History
Introduced in R2023a